
FAQs
1. MVO Portugal
1.1. Who are the members of MVO Portugal?
MVO Portugal was established on 4 August 2017 by the following entities:
- APIFARMA – Associação Portuguesa da Indústria Farmacêutica
- APOGEN – Associação Portuguesa de Medicamentos Genéricos e Biossimilares
- APIEM – Associação Portuguesa de Importadores e Exportadores de Medicamentos
- ADIFA – Associação de Distribuidores Farmacêuticos
- GROQUIFAR – Associação de Grossistas de Produtos Químicos e Farmacêuticos
- AFP – Associação de Farmácias de Portugal
- ANF – Associação Nacional de Farmácias
1.2. What are the responsibilities of MVO Portugal?
MVO Portugal is responsible for the implementation and operation of the national medicines verification system, pursuant to the provisions of Directive 2001/83/EC of the European Parliament and of the Council of 6 November 2001, amended by Directive 2011/62/EU of the European Parliament and of the Council, of 8 June 2011 and by Commission Delegated Regulation (EU) 2016/161 of 2 October 2015. Following the publication of the Delegated Regulation, Decree-Law no. 26/2018, of 24 April was published, transposing the European legislation into the national legal system. More specifically, MVO Portugal’s responsibilities are as follows (article 4 of the Statutes):
-
To apply the requirements set out by EMVO and ensure the general quality of the System’s operation, namely data clean up, the System’s availability and responsiveness and the appropriate security level;
-
To set out the terms and conditions of access to the System, which should be practical and transparent;
-
To set out and implement the necessary requirements and procedures for the regular operation of the System, including the identification and settlement of any irregularities;
-
To hire one or more accredited providers of Information and Communication Technology Services (hereinafter referred to as “ICT Services”) to install and manage the System;
-
To hire the required employees and suppliers to pursue its goal;
-
To prepare and manage the agreements to use the System and set out the provisions regarding remunerations and methods of payment, taking into account article 31 of the Delegated Regulation and what has been agreed among the Permanent Members;
-
To guarantee the communication with the national regulatory authorities on the use of the System to facilitate the procedures of recalling medicinal products and manage other issues regarding the patients’ safety;
-
To prepare and provide the members and the Advisory Board with regular activity reports and statistical reports on the System’s operation and performance;
-
To carry out periodical strategic assessments to the System to guarantee it progresses over time, in the interest of patient safety and in line with the evolution of the healthcare infrastructure in Portugal and in Europe;
-
To invoice and to collect fees, and other participation fees and costs, according to the Directive and the Delegated Regulation and any other amounts owed by the members in terms of the statutes.
1.3. Can MVO Portugal engage in any type of commercial activity?
No. MVO Portugal is not allowed to carry out any commercial activity.
1.4. Is MVO Portugal a new Authority for Medicines?
No. MVO Portugal does not have any responsibility or intervention in the inspective, sanctioning fields, or in other fields that fall within the scope of INFARMED, I.P.’s activity.
1.5. How is MVO Portugal’s activity financed?
MVO Portugal’s activity is financed through the fees paid by the Marketing Authorization or Parallel Import Authorization holders and by its Members (APIFARMA, APOGEN, APIEM, ADIFA, GROQUIFAR, AFP and ANF), according to the model approved by the General Meeting.
2. Safety features
2.1. What are the safety features?
Safety features are the elements to be included in the packaging of the medicinal products covered by the Directive and by the Delegated Regulation that will jointly allow the verification of the authenticity of a medicinal product in an end-to-end verification system.
The safety features to be placed on the packaging are the unique identifier and the anti-tampering device. The verification of the authenticity of the unique identifier aims at ensuring that the medicinal product originates from the legitimate manufacturer. The verification of the integrity of the anti-tampering device shows whether the packaging has been opened or altered since it left the manufacturer, thereby ensuring that the content of the packaging is authentic.
2.2. What is the unique identifier?
The unique identifier consists of the safety feature enabling the verification of the authenticity and the identification of an individual pack of a medicinal product.
2.2.1. What is the composition of the unique identifier for a packaging circulating in Portugal?
The unique identifier is composed of the following elements:
- Product code (GTIN);
- Serial number;
- Batch;
- Expiry date;
- Registration number.
The above mentioned elements will be encoded in a two-dimensional DataMatrix barcode (standard GS1). The five elements mentioned above must be printed on the individual pack. There is no pre-defined order for printing the elements and they can be placed on any side of the individual pack, although it is desirable that, wherever possible, they are placed on the same side of the DataMatrix code. More information can be found here.
2.2.2. What is the composition of the unique identifier for an individual pack circulating in Portugal and Spain?
The unique identifier must contain the following elements:
- Product code (GTIN);
- Serial number;
- Batch;
- Expiry date;
- National registration number – Portugal
- National registration number - Spain
The six elements mentioned above must also be printed on the individual pack. There is no pre-defined order for printing the elements and they can be placed on any side of the individual pack, although it is desirable that, wherever possible, they are placed on the same side of the DataMatrix code. The Spanish national code must be placed on the upper right corner, as is currently required. It should be noted that in Spain it is possible to use GTIN and NTIN. In the case of packaging circulating in Spain and Portugal, the GTIN must always be used, instead of the NTIN.
2.3. What information must be placed on the packaging of the medicinal product in a human readable format?
Article 7 of the Delegated Regulation lays down the elements to be placed on the packaging in human-readable format. In addition, the elements foreseen by the national legislation regarding labelling, namely, those foreseen in article 105 of Decree-Law no. 176/2006 of 30 August, in its current wording, which will continue to be applied without any amendments.
2.4. What is the anti-tampering device?
The anti-tampering device is the safety feature allowing the verification of whether the packaging of a medicinal product has been tampered with.
2.5. What are the specifications the anti-tampering device must comply with?
Neither the Directive nor the Delegated Regulation lay down technical specifications for the anti-tampering device. It is considered that the decisions on specifications will be done by the manufacturers. More information can be found in the document Safety Features for Medicinal Products For Human Use - Questions And Answers;
2.6. What are the medicinal products to which the safety features need to be applied?
According to article 54.º - A of Directive 2001/83/EC, amended by Directive 2011/62/EU, the medicinal products subject to prescription must bear safety features. Annex I of the Delegated Regulation contains the list of medicinal products subject to prescription that shall not bear the safety features:
|
Name of active substance or product category |
Pharmaceutical form |
Strength |
|
Homeopathic medicinal products |
Any |
Any |
|
Radionuclide generators |
Any |
Any |
|
Kits |
Any |
Any |
|
Radionuclide precursors |
Any |
Any |
|
Advanced therapy medicinal products which contain or consist of tissues or cells |
Any |
Any |
|
Medicinal gases |
Medicinal gas |
Any |
|
Solutions for parenteral nutrition having an anatomical therapeutical chemical (‘ATC’) code beginning with B05BA |
Solution for infusion |
Any |
|
Solutions affecting the electrolyte balance having an ATC code beginning with B05BB |
Solution for infusion |
Any |
|
Solutions producing osmotic diuresis having an ATC code beginning with B05BC |
Solution for infusion |
Any |
|
Intravenous solution additives having an ATC code beginning with B05X |
Any |
Any |
|
Solvents and diluting agents, including irrigating solutions, having an ATC code beginning with V07AB |
Any |
Any |
|
Contrast media having an ATC code beginning with V08 |
Any |
Any |
|
Tests for allergic diseases having an ATC code beginning with V04CL |
Any |
Any |
|
Allergen extracts having an ATC code beginning with V01AA |
Any |
Any |
Annex II of the Delegated Regulation contains the list of medicinal products not subject to prescription that shall bear the safety features:
|
Name of active substance or product category |
Pharmaceutical form |
Strength |
|
Omeprazole |
Gastro-resistant capsule, hard |
20 mg |
|
Omeprazole |
Gastro-resistant capsule, hard |
40 mg |
3. The European medicine verification system
3.1. What is the European medicine verification system?
The medicine verification system is a pan-European system to prevent the entry into the legal supply chain of falsified medicinal products and to detect potential falsifications.
The system is composed of a central information and data router (designated as “hub” in the Delegated Regulation and as “EU Hub” in the context of the project) and of repositories which serve the territory of each Member State (designated as “national repositories” in the Delegated Regulation and as “national systems” in the context of the project). The territorial scope of the European medicine verification system is the European Economic Area, composed of the European Union member-states, plus Iceland, Liechtenstein, Norway and Switzerland.
3.2. Who manages the European medicines verification system?
The EU Hub is managed by the European Medicines Verification Organization (EMVO) and each national system is managed by the responsible national organization. An organization counterpart to MVO Portugal has been established in each country involved in the project. The contacts of the several National Medicines Verification Organizations (NMVO) can be found here.
3.3. Access to the EU Hub
The EU Hub is the only data entry point in the entire European medicines verification system. The onboarding partners (OBP) upload into the EU Hub data regarding the packs to be marketed. Only onboarding partners are allowed to upload data to the EU Hub. The process of accessing the EU Hub is called onboarding. More information on the onboarding procedure with the EMVO can be found here.
3.4. What is an onboarding partner?
The onboarding partner (OBP) is the legal entity that will conclude a contract with EMVO to upload data to the EU Hub. This legal entity will act in representation of the companies of the group it belongs to. For example: in the multinational group XPTO that operates in the 32 countries involved in the project, the legal entity XYZ will be the onboarding partner before EMVO and will upload the data regarding the packs that will circulate in the 32 countries. The countries do not need to individually connect to the EU Hub.
3.5. Can the contract manufacturer organizations (CMO) connect directly to the European system?
No. Only MA Holders or Parallel Distributors can conclude a contract with EMVO to upload data, because they are the only entities responsible for the products. The same applies to third party logistics (3PL). More information can be found here.
3.6. When are the OBP able to upload data for Portugal?
The national medicines verification system is operational and in production and is already connected to the EU Hub production environment. Thus, the OBP can already upload the data in the production environment via EU Hub.
3.7. Is the uploading of data to the national repository mandatory, if the repository is operational, but the Delegated Regulation has not yet entered into force?
No. However, it is advisable to upload the data regarding the unique identifiers of the medicinal products released for sale or distribution before the entry into force of the Delegated Regulation, in order to avoid alerts after the entry into force of the Delegated Regulation, due to the lack of information in the system.
3.8. What is the date of entry into force?
The Delegated Regulation will enter into force on 9 February 2019. Thus, all preparatory work must be concluded until that date.
4. The national medicines verification system
4.1. What is the national medicines verification system?
The national medicines verification system is an integral part of the European medicines verification system and consists of the national repository, where the information regarding the packs that circulate in Portuguese territory will be stored. The users of the national system (distributors, pharmacies and healthcare institutions) will establish a connection with it in order to be able to execute their obligations of verification and decommissioning of the unique identifier.
4.2. Was the national medicines verification system created for operations other than the verification and decommissioning of unique identifiers?
No. The system was created to enable the verification and decommissioning of the unique identifiers and does not replace operations that already exist in the users’ organizations. For example, the system was not created for stock management, financial management and other procedures.
4.3. How does the system work?
Schematically and summarily, the system will operate as follows: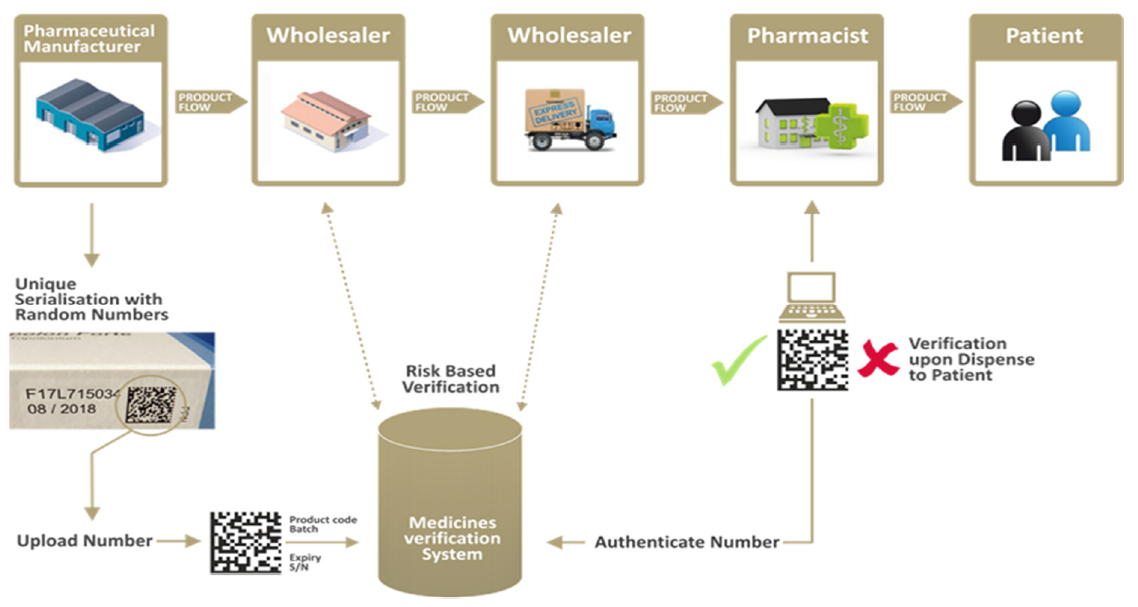
Fonte: EMVO.
The verification of the authenticity of the medicinal product is performed by comparing the information regarding the unique identifier, registered by the OBP, with the information regarding the unique identifier that is placed on the pack.
The data regarding each pack will be uploaded to the system by the onboarding partner (the entity responsible before EMVO for uploading the data) through the EU Hub. In turn, the EU Hub will forward the data, in automated form, to the relevant national systems, where it will reside. The users (distributors, pharmacies and healthcare institutions), through the connection established for that purpose, will scan the unique identifier placed on the pack in order to compare it with the information contained in the national repository.
The verification of the anti-tampering device is performed by means of visual inspection and, therefore, without resorting to the system.
4.4. Will a pack be verified in all points of the supply chain it passes through?
Not necessarily, because the system was not conceived in a track & trace logic. The system was conceived in accordance with a risk-based logic, meaning that there are points of the supply chain where the verification is not mandatory, because the risk of falsification is considered negligible.
Article 20(b) of the Delegated Regulation mentions that the wholesaler (for the purposes of this document, wholesaler and distributor shall be regarded as synonyms) shall verify the “medicinal products he receives from a wholesaler who is neither the manufacturer nor the wholesaler holding the marketing authorization nor a wholesaler who is designated by the marketing authorization holder, by means of a written contract, to store and distribute the products covered by his marketing authorization on his behalf”. This means that the Distributor is not required to verify the medicinal products he receives from the manufacturer, from the wholesaler holding the marketing authorization or from a wholesaler who is designated by the marketing authorization holder, by means of a written agreement, to store and distribute the products covered by his marketing authorization on his behalf.
It should be noted that the entities at the ends of the legal supply chain (onboarding partners, on one hand, and persons authorized or entitled to supply medicines to the public, on the other hand) are always obliged to perform the operations defined in the Directive and in the Delegated Regulation, and the risk-based verification logic does not apply.
4.5. Is there an obligation of verification of the packs in the inverse logistic?
Yes. As mentioned in item b) of article 22 of the Delegated Regulation, the wholesaler shall verify the authenticity of the medicinal products “which have been returned to him by persons authorized or entitled to supply medicinal products to the public or another wholesaler and cannot be returned to saleable stock”.
It should be noted that the Delegated Regulation does not make any reference to the reason for return. Such as, for example, packs returned for destruction after they have expired must be verified.
4.6. Who has access to what data on the system?
According to article 38 of the Delegated Regulation, “manufacturers, marketing authorization holders, wholesalers and persons authorized or entitled to supply medicinal products to the public shall be responsible for any data generated when they interact with the repositories system and stored in the audit trail. They shall only have ownership of and access to those data, with the exception of the information referred to in Article 33(2) and the information on the status of a unique identifier”.
MVO Portugal cannot have access to the audit trail and to the data contained therein without the written agreement of the legitimate data owners except for the purpose of investigating potential incidents of falsification flagged in the system in accordance with Article 36(b) of the Delegated Regulation.
INFARMED, in the capacity of National Authority for Medicinal Products, shall have access to the national medicines verification system, for the following purposes:
-
supervising the functioning of the repositories and investigating potential incidents of falsification;
-
reimbursement;
-
pharmacovigilance or pharmacoepidemiology.
4.7. What is the audit trail?
According to article 35(g) of the Delegated Regulation, the audit trail is the complete record of all operations concerning a unique identifier, of the users performing those operations and the nature of the operations. The audit trail shall be created when the unique identifier is uploaded to the repository and be maintained until at least one year after the expiry date of the medicinal product bearing the unique identifier or five years after the product has been released for sale or distribution in accordance with Article 51(3) of Directive 2001/83/EC, whichever is the longer period.
4.8. What happens when the data of the unique identifier does not coincide with the data registered in the repository?
Regarding a specific pack, if the data of the unique identifier does not coincide with the data registered in the repository, the system will trigger a potential falsification alert (see article 36(b) of the Delegated Regulation).
4.9. What to do in the event of potential falsification alert?
In the event of a potential falsification alert, proceed according to the documentation available here.
4.10. How shall the alerts triggered before 9 February 2019 be treated?
According to INFARMED, I.P.’s interpretation the alerts triggered by the system before 9 February 2019 have no legal value, because they will occur before the entry into force of the Delegated Regulation. The alerts triggered before the entry into force of the Delegated Regulation will not result in any formal procedure.
4.11. Is the implementation of the system or its operation different in the Autonomous Regions?
No. All rules are fully applicable to the entities with head-office, or operating, in the Autonomous Regions.
4.12. What is the relationship between the financial procedures and the verification and decommissioning operations?
The operations of verification and decommissioning of unique identifiers are independent from the financial procedures associated with the medicinal products concerned.
4.13. Will the aggregation of unique identifiers be implemented?
The Delegated Regulation neither mandates nor prohibits the implementation of aggregation solutions. Discussions are currently in progress at EU level concerning possible scenarios for the implementation of aggregation solutions, namely, within the context of healthcare institutions. To date, no final and binding decisions have been taken with regard to the implementation of aggregation solutions within the context of the European medicines verification system.
5. Access to the national medicines verification system
5.1. Who is obliged to connect with the national medicines verification system?
The following entities, pursuing their activity in national territory, are obliged to connect to the national medicines verification system:
- o Distributors: holders of a valid wholesale distribution authorization issued by INFARMED, I.P. or by the competent authority in the Autonomous Regions;
- o Community pharmacies: holders of a valid license, issued by INFARMED, I.P. or by the competent authority in the Autonomous Regions;
- o Healthcare institutions: holders of a license for the direct procurement of medicinal products, issued by the INFARMED, I.P. or by the competent authority in the Autonomous Regions, providing outpatient or inpatient treatment.
5.2. Will it be possible to share access credentials?
No. The national medicines verification system’s access credentials are granted by MVO Portugal to each entity after the successful completion of the validation process (referred to as onboarding process). The access credentials are not transferable and each user shall perform his activities in the system using the credentials granted to him by MVO Portugal. For example, a distributor shall perform all his activities with the access credentials granted to him by MVO Portugal, independently of the activities he performs on the system or of the proprietor of the medicinal products he possesses.
5.3. What are the consequences for an entity that does not connect to the system?
A company that is required to perform operations of verification and decommissioning of unique identifiers and fails to connect to the national medicines verification system will not be able to fulfill its legal obligations in this respect. The applicable penalties will be defined and executed by the competent authorities.
Each entity is responsible for guaranteeing the necessary preparation for the access to the national medicines system.
5.4. What healthcare institutions should be connected to the national medicines verification system?
According to the provisions of article 105-A(9) of Decree-Law no. 26/2018 of 24 April, the establishments and services holding a license for the direct procurement of medicinal products issued by INFARMED, I.P., that are integrated in public or private healthcare institutions, that provide medical and pharmaceutical services and inpatient treatment, are obliged to be connected.
5.5. What entities are not obliged to connect to the national medicines verification system?
The entities referred to in article 23 of the Delegated Regulation and the entities referred to in article 105-A(9) of Decree-Law no. 26/2018 of 24 April are not obliged to connect to the national medicines verification system.
5.6. Do MA Holders or parallel import authorization holders have access to the national medicines verification system?
No. Marketing authorization holders and parallel import authorization holders will perform the operations in the system through the EU Hub.
Marketing authorization holders and parallel import authorization holders in possession of a wholesale distribution authorization for medicinal products for human use issued by INFARMED, I.P. and distributing their medicinal products or the medicinal products they represent must be connected to the national medicines verification system. For this purpose, they shall be considered as distributors and must follow the onboarding process set forth.
5.7. What is the validation procedure for accessing the national medicines verification system?
In order to comply with the obligations arising from the legislation, namely, with regard to the verification and decommissioning of the packs in their possession, distributors, pharmacies and healthcare institutions must connect their information systems to the national medicines verification system.
For a user to access the national medicines verification system, the access validation process must be successfully completed. This process is called onboarding process and its purposes are the validation of the legitimacy to access the system, the technical validation and the signature of the agreement governing the relationship between each entity and MVO Portugal. The decisions concerning the technical integration scenario (e.g., the software to be used, external supply vs. internal development, etc.) are taken by each entity. Likewise, the decisions concerning the organization and the internal procedures are the exclusive responsibility of each entity.
- The onboarding process for distributors can be found here.
- The onboarding process for pharmacies can be found here.
- The onboarding process for healthcare institutions can be found here.
5.8. Who must ensure the onboarding process?
With respect to the distributors and the healthcare institutions, each entity is responsible for ensuring that the onboarding process is successfully concluded. With respect to the community pharmacies, the process will be ensured by the Pharmacies Associations (AFP and ANF). The pharmacies that are not members of the AFP or of the ANF, may choose to connect to the national system through one of the Associations or through direct connection to the national system. For more information, please contact AFP through the website www.afp.com.pt, ANF through the website www.anf.pt, or MVO Portugal through the email address mvo.portugal@mvoportugal.pt.
5.9. What is the pilot phase and how is it structured?
The pilot phase consists of a period of tests the main purpose of which is to use the system in conditions as close as possible to the real conditions in which the system will be used after the entry into force of the Delegated Regulation. For the execution of this set of tests, the participation of MA Holders or of Parallel Distributors, distributors, pharmacies and healthcare institutions is necessary, for each of them to perform their daily verification and unique identifier decommissioning tasks and to ensure that it is possible to verify the flux of packs from the manufacturer to the point of dispense. The following aspects should be highlighted:
- o The participation in the pilot phase is voluntary. There is no obligation to participate nor any penalty for lack of participation;
- o The focus of the tests to be made shall reside in the interaction capability between the systems. The aspects regarding internal organization and procedures are the exclusive responsibility of each participant and shall not be verified by MVO Portugal;
- o Different participants may be in conditions to participate in different moments. Thus, a maximum of flexibility is intended: the date of entry into the pilot stage is decided by the participant himself and each participant may execute the tests without depending on other participants;
- o The execution of the tests is previously planned with MVO Portugal;
- o The test scenarios to be executed are defined by MVO Portugal and oriented according to each type of participant (MA Holder/Parallel Distributor, Distributor, Pharmacy, Healthcare Institution). The participants may add other test scenarios they consider relevant. Additional test scenarios will not be accompanied by MVO Portugal.
- o For the execution of tests in the test environment (IQE), the AM Holders/Parallel Distributors must use their own data and the other participants will use data provided by MVO Portugal or by AM Holders/Parallel Distributors;
- o It is considered that all operations performed in the production environment (PRD) are real transactions, therefore, the monitoring of the activity will depend on the establishment of the several participants’ connection to the system.
The participants in the pilot phase must document the results of the tests using the form to be provided for this purpose. This information is necessary for the attribution of the access credentials to the production environment.
6. Transition Period
6.1. Is a transition period foreseen?
Yes. According to article 48 of the Delegated Regulation, “medicinal products that have been released for sale or distribution without the safety features in a Member State before the date in which this Regulation becomes applicable in that Member State, and are not repackaged or relabelled thereafter, may be placed on the market, distributed and supplied to the public in that Member State until their expiry date”.
6.2. Is the coexistence of packs in the market with the barcode 39 and with the datamatrix code possible?
Yes, provided that the barcode 39 does not have impact on the readability of the outer package.
6.3. Is the removal of the barcode 39 mandatory for batches released from 9 February 2019 onwards?
No.
7. Where can more information be found
More information can be found in the following documents:
If you did not find the answer to your question, please send your question to mvo.portugal@mvoportugal.pt.



